Taxonomic treatments
For a demo for Donat Agosti, I've added some taxonomic treatments to iSpecies.org. I'd done this before, but after the server got hacked I didn't restore the treatments because they are served via a triple store, and I hadn't got that running. Now that the triple store is up, I looked at this again. Here's what is involved.
Treatments
TaxonX is a XML mark up for taxonomic descriptions. The idea is to locate and mark blocks of text that describe a taxon. For more details see the AMNH's NSF Taxonomic Literature Projectpages.
Donat has been marking up various ant papers manually as proof of concept, but the process will be automated. I want to be able to serve up a taxonomic description of a name, e.g. "Proceratium google". Because I want everything to be in a triple store, I need to map TaxonX to RDF. Here's what I do:
So, a publication is modelled like this:

And a treatment is modelled like this.

SPARQL
Currently iSpecies treatments are retrieved using RDQL, but SPARQL is rather nicer. Finding the treatment for a taxon is a simple SPARQL query, e.g.:
Display
To display the results I take the SPARQL XML result, convert the encoded TaxonX block to XML mark up, then apply a simple XSLT style sheet. The results aren't pretty, but it works.
Future directions
As I've mentioned in an earlier post, what I'd really like is to have GUIDs for these publications sorted out, and more mark up. In particular, literature cited, specimens, and other taxonomic names should be marked up so that these links can be extracted. If this is done well, then we could do things like:
The trick will transforming TaxonX to RDF.
Currently playing in iTunes: Shelter by Ray LaMontagne
Treatments
TaxonX is a XML mark up for taxonomic descriptions. The idea is to locate and mark blocks of text that describe a taxon. For more details see the AMNH's NSF Taxonomic Literature Projectpages.
Donat has been marking up various ant papers manually as proof of concept, but the process will be automated. I want to be able to serve up a taxonomic description of a name, e.g. "Proceratium google". Because I want everything to be in a triple store, I need to map TaxonX to RDF. Here's what I do:
- The URI of the paper is the link to the PDF in AntBase. This should really be something else (LSID, DOI, Handle, PURL), but it will do for now.
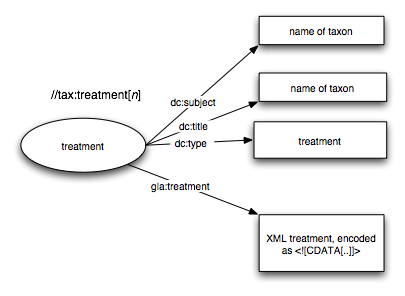
- Each treatment is extracted from the TaxonX document using XPath. I use a Perl script to pull out each node matching //tax:treatment
- Each treatment gets a URI, based on the URI of the paper containing the treatment, and the XPath to the treatment, e.g. http://antbase.org/ants/publications/8538_fisher//tax:treatment[1]. The idea is that one could use the identifier to extract the relevant block of text from the TaxonX XML document (i.e., the identifier would be useful beyond my triple store). Although I worry that this is not semantically opaque, its seems a useful idea, and my worries eased when I discovered that Annotea uses the same idea.
- The actual treatment is stored as a block of <![CDATA[..]]>, so the original TaxonX markup is preserved.
- Each treatment is linked to the containing paper by the Dublin core term <dcterms:isPartOf>. I also have the inverse link <dcterms:hasPart> to link the publication to the treatments it contains.
- I have some minimal metadata about the publication (title, format), and about each treatment (name of taxon stored in <dc:subject>). This is extracted from what is in the TaxonX document - clear TaxonX needs more information on the source.
- Each treatment is typed using <dc:type>treatment<dc:type>. I do this so that I can classify results for a query (as part of another project).
So, a publication is modelled like this:

And a treatment is modelled like this.
SPARQL
Currently iSpecies treatments are retrieved using RDQL, but SPARQL is rather nicer. Finding the treatment for a taxon is a simple SPARQL query, e.g.:
PREFIX gla: <urn:lsid:lsid.zoology.gla.ac.uk:predicates:>
PREFIX dc: <http://purl.org/dc/elements/1.1/>
PREFIX dcterms: <http://purl.org/dc/terms/>
PREFIX tax: <http://research.amnh.org/informatics/taxlit/taxonx/taxonx1>
SELECT ?uri ?publication ?title ?treatment
WHERE {?uri dc:subject 'Proceratium google'
?uri gla:treatment ?treatment .
?publication dcterms:hasPart ?uri .
?publication dc:title ?title
}
Display
To display the results I take the SPARQL XML result, convert the encoded TaxonX block to XML mark up, then apply a simple XSLT style sheet. The results aren't pretty, but it works.
Future directions
As I've mentioned in an earlier post, what I'd really like is to have GUIDs for these publications sorted out, and more mark up. In particular, literature cited, specimens, and other taxonomic names should be marked up so that these links can be extracted. If this is done well, then we could do things like:
- Generate distribution maps for papers that don't have maps
- Generate synonymies from lists of names
- Infer type status even if specimen databases don't have this information
- etc.
The trick will transforming TaxonX to RDF.
Currently playing in iTunes: Shelter by Ray LaMontagne
posted by Roderic Page at
Sunday, July 16, 2006
|
0 comments
![]()

 Now, in an ideal, joined-up world, we'd have a link from the Fisher paper to the actual specimen. A bit of fussing (i.e., searching for "Strumigenys rubigus" on AntWeb) reveals that the holotype is
Now, in an ideal, joined-up world, we'd have a link from the Fisher paper to the actual specimen. A bit of fussing (i.e., searching for "Strumigenys rubigus" on AntWeb) reveals that the holotype is  to get the tree effect.
to get the tree effect.
